The Answers You Need
Trusted healthcare information from our experts

At GoodRx Health, we’re serious about getting you health information you can trust. Every article is edited and reviewed by experts, so you can feel confident in what you’re reading — and how you use it.
Learn more
Our content is honest and unbiased, focused on clear and comprehensive health information.

Accuracy comes first. Our content is carefully researched and medically reviewed by qualified professionals.

Health isn’t one-size-fits-all. We explain things clearly and thoughtfully, so you can make decisions that feel right for you.




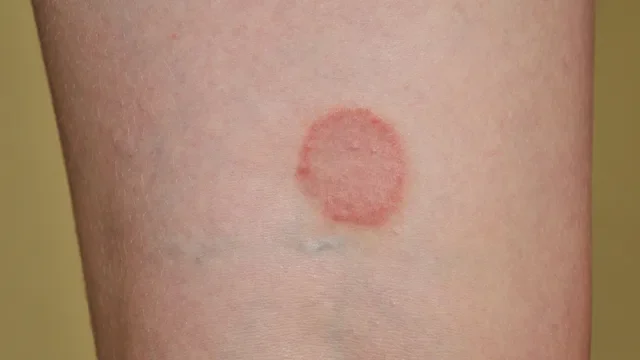
Head-to-head comparisons of medication uses, side effects, warnings, and more.
Learn how




Find out if Medicare Advantage or Medigap is best.
Take the quiz
Brand names at an affordable price, plus unlimited online care.
Learn more





Prescription savings
Stop paying too much for your prescriptions. Compare prices, get pharmacy coupons, and save up to 80%.Resources
About GoodRx
Health conditions
Medications & treatment
Access & affordability
Well-being
Medication discounts